Ein Gen steuert Entwicklung von Gehirn und Schädel
Eine neue Studie liefert eine Erklärung dafür, warum bestimmte Leukämiepatienten nicht auf eine Therapie ansprechen
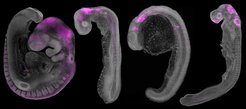
 in Embryonen von vier verschiedenen Wirbeltierarten mittels HCR in situ-Hybridisierung. Von links nach rechts: Maus, Huhn, Zebrafisch und Katzenhai.")
Auf den Punkt gebracht
- Ursprung in der Urzeit: MN1 ist ein uraltes Gen, das sich beim Übergang zu Wirbeltieren strukturell veränderte.
- Integration: MN1 wurde in frühzeitliche molekulare Mechanismen integriert und trieb neue Entwicklungsprozesse an.
- Verbindung zwischen Gehirn und Schädel: MN1 steuert sowohl die Musterbildung des Gehirns als auch die Schädelbildung – und liefert damit erstmals den genetischen und mechanistischen Nachweis dafür, wie Gehirn und Schädel gemeinsam entstanden und sich entwickelten.
- Medizinische Relevanz: Wer MN1 versteht, kann angeborene Syndrome besser begreifen – und auch, warum manche Leukämiepatienten nicht auf bestimmte Therapien ansprechen.
Das Gehirn und der Schädel sind ein biologisches Duo – sie wachsen und formen sich in perfekter Koordination während der frühen Entwicklung. Diese enge Partnerschaft ist kein Zufall. Im Laufe von Millionen Jahren von Evolution haben sich Gehirn und Schädel gemeinsam angepasst und gegenseitig geformt, um Schutz, Funktion und Überleben zu gewährleisten. Forschende beobachten diesen Zusammenhang seit langem, aber die genetischen Anweisungen, die dieses synchronisierte Wachstum steuern – und wie es sich entwickelt hat – blieben weitestgehend unbekannt. Bis jetzt.
Forschende des Max-Planck-Instituts für Evolutionsbiologie unter der Leitung von Markéta Kaucká haben entdeckt, dass MN1, ein Gen, das ursprünglich mit Hirntumoren und Leukämie assoziiert wurde, sich tatsächlich bereits vor Hunderten von Millionen Jahren in primitiven wirbellosen Tieren entwickelt hat. Mit dem Aufkommen der Wirbeltiere, die über komplexere Gehirne und Schädel verfügen, erfuhr das Gen strukturelle Veränderungen und wurde für die Entwicklung der Wirbeltiere unverzichtbar.
Ein Gen aus der Frühzeit der Tierentwicklung
 in einem Maus-Embryo am 10. Embryonaltag mittels HCR in situ-Hybridisierung.")
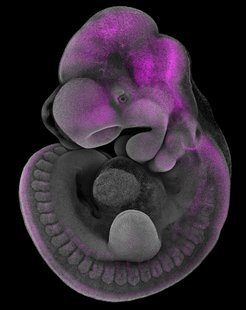
Die Forschenden verfolgten die Abstammung von MN1 bis zu primitiven wirbellosen Tieren zurück. Obwohl es sich strukturell von dem in wirbellosen Tieren unterscheidet, blieb die Kernsequenz von MN1 während der Evolution der Wirbeltiere erhalten. In kiefertragenden Wirbeltieren erhielt MN1 ein neues kurzes Exon, das eine C-terminale Domäne kodiert, die für seine Funktion bei der Gehirnentwicklung und Schädelbildung unerlässlich wurde.
Das Team fand heraus, dass MN1 bei der frühen Entwicklung zur Musterbildung des embryonalen Gehirns beiträgt und damit die Bildung der Schädelknochen steuert. Ohne MN1 ist die Segmentierung des Gehirns beeinträchtigt, die Entwicklung der Hirnnerven ist abnormal und die Schädelknochen entwickeln sich fehlerhaft – ähnlich wie bei menschlichen Syndromen, die sich durch Gaumenspalten, Schädeldeformitäten und neurologischen Entwicklungsverzögerungen äußern.
Bemerkenswerterweise spiegeln diese Defekte die zuvor beobachteten Auswirkungen eines veränderten Retinsäurespiegels wider. Das Team konnte nachweisen, dass MN1 den Gehalt an Retinsäure, einem für die Embryonalentwicklung essentiellen Molekül, sowie die Expressionsmuster von Hox-Genen, die wie ein Bauplan fungieren, der den Körperbau eines Embryos entlang der Kopf-Schwanz-Achse steuert, kontrolliert und brachte somit die Funktion von MN1 mit einem bekannten und uralten Signalweg in Verbindung.
Synchrones Wachstum
Die Entdeckung gibt Aufschluss darüber, wie sich bei Wirbeltieren die beiden charakteristischen Merkmale – Gehirn und Schädel – entwickelt haben und wie ein einzelnes Gen dazu beiträgt, deren Bildung und Wachstum zu synchronisieren. Die Forschung beleuchtet auch ein umfassenderes Konzept: wie die Evolution von Genen deren Integration in uralte molekulare Systeme ermöglicht und Innovationen und makroevolutionäre Übergänge vorantreibt.
Darüber hinaus bieten die Ergebnisse der Studie eine Erklärung dafür, warum bestimmte Leukämiepatienten nicht auf eine Retinsäure-Therapie ansprechen. Patienten mit einem hohen MN1-Spiegel weisen eine Therapieresistenz auf, die vermutlich darauf zurückzuführen ist, dass MN1 einen schnellen Abbau des Medikaments ermöglicht und so dessen Wirkung verhindert. Die Studie zeigt zudem Verbindungen zu zahlreichen menschlichen Erkrankungen auf, bei denen Verkürzungen und Mutationen in MN1 mit neurologischen Entwicklungsstörungen und kraniofazialen Syndromen assoziiert wurden.











